Prawo Hardy’ego Weinberga i model dryfu genetycznego
Podstawowe pojęcia związane z dziedziczeniem
Dziedziczenie w populacji wirtualnej można zaprogramować poprawnie tylko na jeden sposób: naśladując przyrodę. Może to być klonowanie – dziedziczenie wszystkich genów od osobnika macierzystego lub rozród generatywny – dosłowne odtworzenie zasad dziedziczenia opisanych przez Gregora Mendla. Obecne ćwiczenie dotyczyć będzie rozrodu generatywnego.
Na początek należy przyswoić sobie znaczenie pewnych terminów (wprowadzanych jeszcze w szkole, ale często mylonych)
- Gen – odcinek DNA podlegający transkrypcji (czyli “przepisywany” za pomocą odpowiedniej reakcji chemicznej) na pewien typ RNA (mRNA, tRNA, rRNA i in.), a pośrednio kodujący zwykle także jakieś białko. Dawniej termin gen oznaczał podstawową jednostkę dziedziczenia określonej cechy organizmu (np. koloru oczu). Czasem jeszcze taka definicja funkcjonuje w pracach biologicznych.
- Locus – fragment DNA zajmujący określone miejsce w cząsteczce kwasu nukleinowego. Może być to gen we współczesnym znaczeniu albo fragment DNA nie podlegający transkrypcji umiejscowiony w okolicach określonych genów.
- Allel – wersje tego samego locus (np. genu) różniące się od siebie sekwencją nukleotydów. Choć pod tą definicję można podciągnąć wszystkie różne fragmenty DNA – to w rzeczywistości w tym samym locus na tysiące (setki tysięcy) par zasad nukleotydowych obserwuje się co najwyżej kilka-kilkanaście różnic w ich sekwencji. Allele nie są fragmentami DNA tylko nazwami możliwych DNA związanych z tym samym locus. Zwyczajowo nazwy te mają charakter umowny (np. A i a, albo a i b, albo A1, A2, A3,…). Kilkaset tysięcy różnych nukleotydów DNA tworzących ten sam locus może mieć tylko kilka różnych alleli albo nawet tylko 1 allel.
- Genotyp – pojęcie związane tylko z jednym genem i pokazujące jakie allele tego genu występują w danym osobniku (np. aA, AB – genotyp osobnika heterozygotycznego lub AA – genotyp osobnika homozygotycznego).
- Genom – wszystkie (ale liczone tylko raz) geny występujące u danego osobnika. Zatem osobniki diploidalne mają dwa genomy, tetraploidalne (jak pszenżyto) mają 4 genomy itd.
Kolejne pojęcia związane są opisem struktury genowej całej populacji:
- Pula genowa – geny związane z tym samym locus wszystkich osobników należących do populacji. W populacji osobników diploidalnych liczebność puli genowej jest dwa razy większa niż liczebność populacji.
- Struktura genetyczna (genowa) populacji – udziały procentowe genów określonego allelu w puli genowej populacji.
- Populacja monomorficzna – populacja, u której wszystkie geny w puli genowej mają jeden i ten sam allel.
- Populacja polimorficzna – populacja, u której w puli genowej można wyróżnić różne (co najmniej dwa) allele.
Pojęcia te dotyczą stanu populacji w określonym momencie. Tak jak liczebność populacji może zmieniać się w czasie, tak zmienia się struktura genetyczna populacji. Populacja polimorficzna może stać się monomorficzna, a przy mutacjach genu może zajść zjawisko odwrotne. Populacje wirtualne bardzo często służą do ilustrowania zmian w czasie struktury genowej. Takie modele powstają jako proste (choć niektórzy twierdzą, że pracochłonne – średnio 20 minut uważnej pracy) modyfikacje wykonywanych do tej pory programów poprzez wprowadzenie do rekordu opisującego osobnika zmiennej gen (osobniki haploidalne) lub pary genów: gen1, gen2 (osobniki diploidalne) oraz programowanie dziedziczenia.
Najczęstsze strategie dziedziczenia genów
Nie ma żyjących osobników bez nośników informacji o budowie i funkcjonowaniu ciała osobników. Nośniki te nazwano kiedyś genami i obecnie wiemy że są to fragmenty nici DNA (choć nie koniecznie tylko te podlegające transkrypcji). Są one dziedziczone przez potomków. Mówi się czasem o przekazywaniu cech potomkom. W rzeczywistości rozród jest zawsze kopiowaniem nici DNA osobników rodzicielskich i przekazywanie ich potomkom. Najczęstszymi strategiami dziedziczenia jest klonowanie i rozród generatywny, który jest nieco odmienny w populacjach nierozdzielnopłciowych i rozdzielnopłciowych.
- klonowanie – dziedziczenie genu lub genotypu w całości od osobnika macierzystego. Taki schemat odpowiada wielu mikroorganizmom i osobnikom rozmnażającym się wyłącznie lub prawie wyłącznie wegetatywnie.
- rozród generatywny w populacji nierozdzielnopłciowej – dziedziczenie losowo wybranego genu od osobnika macierzystego i losowo wybranego genu od losowego wybranego osobnika całej populacji. Taki schemat dziedziczenia jest odpowiedni dla modeli większości populacji roślinnych. Ten rodzaj dziedziczenia nazywany jest powszechnie dziedziczeniem Mendlowskim.
- Rozród generatywny w populacji rozdzielnopłciowej – dziedziczenie losowo wybranego genu od osobnika macierzystego i losowo wybranego genu od losowego wybranego osobnika ojcowskiego. Taki schemat dziedziczenia jest odpowiedni dla modeli większości populacji zwierzęcych. W modelu wymaga to określenia płci u osobników i dopuszczenia do rozrodu tylko osobniki żeńskie. Osobniki męskie są wtedy wyłącznie dawcami genów.
W najprostszych modelach geny nie wpływają na mechanizm regulacyjny populacji, nie zmieniają prawdopodobieństw rozrodu, śmierci i liczbę potomków osobnika. Są neutralne. Założenia tego modelu są dokładnie takie same jak założenia w prawie Hardy’ego Weinberga. Poza jednym: modelowane populacje nie muszą być bardzo duże.
Kody źródłowe programów symulujących populacje z trzema strategiami dziedziczenia
Dziedziczenia nie da się zaprogramować w modelu populacji bez definiowania osobników. Dlatego też pokazane programy są obiektowymi modelami populacji. Osobniki nie ograniczają się już do indywidualnego numeru, ale posiadają nośniki genów, tradycyjnie w literaturze, zwłaszcza modelarskiej, nazywane genami. W populacji o rozrodzie klonalnym wystarczy 1 gen, choć taki rozród jest dość częsty w populacjach osobników diploidalnych. Pozostałe strategie rozrodu realizowane są wyłącznie u osobników diploidalnych.
Kod źródłowy programu kodującego populację o dziedziczeniu klonalnym:
from random import random
from math import exp
class stanosobnika:
NR=0
GEN=''
populacja=[]
populacja1=[]
ar=-0.005
br=1
ass=0.005
bs=0
N0=int(-(br-bs)/(ar-ass))
czas=500
lpow=5
tekst='Powtorzenie, Czas, N, NA, NB \n'
wyniki = open('wyniki.txt', 'w')
for pow in range(1,lpow+1):
nA=0
nB=0
ostnr=1
for i in range(N0):
osob=stanosobnika()
osob.NR=ostnr
ostnr=ostnr+1
if random()<0.5 :
osob.GEN='A'
nA=nA+1
else:
osob.GEN='B'
nB=nB+1
populacja.append(osob)
N=N0
t=0
tekst=tekst+str(pow)+", "+str(t)+", "+str(N)+", "+str(nA)+", "+str(nB)+"\n"
for t in range(1,czas+1):
n=0
pr=1/(1+exp(-(ar*N+br)))
ps=1/(1+exp(-(ass*N+bs)))
for i in range(N):
osob=populacja[i]
if random()<pr:
potom=stanosobnika()
potom.NR=ostnr+1
ostnr=ostnr+1
potom.GEN=osob.GEN
populacja1.append(potom)
n=n+1
if random()<ps:
pass
else:
populacja1.append(osob)
n=n+1
N=n
if N>100000:
break
nA=0
nB=0
for i in range(N):
osob=populacja1[i]
if osob.GEN=='A':
nA=nA+1
else:
nB=nB+1
populacja=populacja1
populacja1=[]
tekst=tekst+str(pow)+", "+str(t)+", "+str(N)+", "+str(nA)+", "+str(nB)+"\n"
populacja = []
open('wyniki.txt', 'a').write(tekst)
wyniki.close()
|
Kod źródłowy programu kodującego populację o dziedziczeniu generatywnym dla populacji nierozdzielnopłciowych:
from random import random, randrange
from math import exp
class stanosobnika:
NR=0
GEN1=''
GEN2=''
populacja=[]
populacja1=[]
N0=100
ar=-0.005
br=1
ass=0.005
bs=0
czas=500
lpow=5
tekst='Powtorzenie, Czas, N, NAA, NAB, NBB, NA, NB \n'
wyniki = open('wyniki.txt', 'w')
for pow in range(1,lpow+1):
nA=0
nB=0
nAA=0
nBB=0
nAB=0
for i in range(N0):
osob=stanosobnika()
osob.NR=i
if i<N0/2:
osob.GEN1='A'
nA=nA+1
else:
osob.GEN1='B'
nB=nB+1
if (i<N0/4 or i>=3*N0/4):
osob.GEN2='A'
nA=nA+1
else:
osob.GEN2='B'
nB=nB+1
if (osob.GEN1=='A' and osob.GEN2=="A"): nAA=nAA+1
elif (osob.GEN1=="B" and osob.GEN2=="B"): nBB=nBB+1
else: nAB=nAB+1
populacja.append(osob)
N=N0
ostnr=N0
t=0
tekst=tekst+str(pow)+", "+str(t)+", "+str(N)+", "+str(nAA)+", "+str(nAB)+", "+str(nBB)
tekst=tekst+", "+str(nA)+", "+ str(nB)+"\n"
for t in range(1,czas+1):
n=0
pr=1/(1+exp(-(ar*N+br)))
ps=1/(1+exp(-(ass*N+bs)))
for i in range(N):
osob=populacja[i]
if random()<pr:
potom=stanosobnika()
potom.NR=ostnr+1
ostnr=ostnr+1
if random()<0.5:
potom.GEN1=osob.GEN1
else:
potom.GEN1=osob.GEN2
ojciec=populacja[randrange(N)]
if random()<0.5:
potom.GEN2=ojciec.GEN1
else:
potom.GEN2=ojciec.GEN2
populacja1.append(potom)
n=n+1
if random()<ps: pass
else:
populacja1.append(osob)
n=n+1
N=n
if N>100000:
break
nAA=0
nBB=0
nAB=0
nA=0
nB=0
for i in range(N):
osob=populacja1[i]
if osob.GEN1=='A': nA=nA+1
else: nB=nB+1
if osob.GEN2=='A': nA=nA+1
else: nB=nB+1
if (osob.GEN1=='A' and osob.GEN2=="A"): nAA=nAA+1
elif (osob.GEN1=="B" and osob.GEN2=="B"): nBB=nBB+1
else: nAB=nAB+1
populacja=populacja1
populacja1=[]
tekst=tekst+str(pow)+", "+str(t)+", "+str(N)+", "+str(nAA)+", "+str(nAB)+", "+str(nBB)+", "+str(nA)
tekst=tekst+", "+str(nB)+"\n"
populacja=[]
open('wyniki.txt', 'a').write(tekst)
wyniki.close()
|
Kod źródłowy programu kodującego populację o dziedziczeniu generatywnym dla populacji rozdzielnopłciowych:
from random import random, randrange
from math import exp
class stanosobnika:
NR=0
PLEC=''
GEN1=''
GEN2=''
populacja=[]
populacja1=[]
N0=100
ar=-0.01
br=1
ass=0.005
bs=-1.5
czas=500
lpow=5
tekst='Powtorzenie, Czas, N, Nf, Nm, NfAA, NfAB, NfBB, NmAA, NmAB, NmBB, nfA, nfB, nmA, nmB \n'
wyniki = open('wyniki.txt', 'w')
for pow in range(1,lpow+1):
nfA=0
nfB=0
nmA=0
nmB=0
NfAA=0
NfAB=0
NfBB=0
NmAA=0
NmAB=0
NmBB=0
for i in range(N0):
osob=stanosobnika()
osob.NR=i
if random()<0.5: osob.PLEC='F'
else: osob.PLEC='M'
if (random()<0.5):
osob.GEN1='A'
else:
osob.GEN1='B'
if (random()<0.5):
osob.GEN2='A'
else:
osob.GEN2='B'
if (osob.PLEC=='F' and osob.GEN1=='A' and osob.GEN2=='A'):
NfAA=NfAA+1
nfA=nfA+2
if (osob.PLEC=='F' and osob.GEN1=='A' and osob.GEN2=='B'):
NfAB=NfAB+1
nfA=nfA+1
nfB=nfB+1
if (osob.PLEC=='F' and osob.GEN1=='B' and osob.GEN2=='A'):
NfAB=NfAB+1
nfA=nfA+1
nfB=nfB+1
if (osob.PLEC=='F' and osob.GEN1=='B' and osob.GEN2=='B'):
NfBB=NfBB+1
nfB=nfB+2
if (osob.PLEC=='M' and osob.GEN1=='A' and osob.GEN2=='A'):
NmAA=NmAA+1
nmA=nmA+2
if (osob.PLEC=='M' and osob.GEN1=='A' and osob.GEN2=='B'):
NmAB=NmAB+1
nmA=nmA+1
nmB=nmB+1
if (osob.PLEC=='M' and osob.GEN1=='B' and osob.GEN2=='A'):
NmAB=NmAB+1
nmA=nmA+1
nmB=nmB+1
if (osob.PLEC=='M' and osob.GEN1=='B' and osob.GEN2=='B'):
NmBB=NmBB+1
nmB=nmB+2
populacja.append(osob)
N=N0
ostnr=N0
t=0
Nf=NfAA+NfAB+NfBB
Nm=NmAA+NmAB+NmBB
tekst=tekst+str(pow)+", "+str(t)+", "+str(N)+", "+str(Nf)+", "+str(Nm)
tekst=tekst+", "+str(NfAA)+", "+str(NfAB)+", "+str(NfBB)+", "+str(NmAA)+", "+str(NmAB)
tekst=tekst+", "+str(NmBB)+", "+str(nfA)+", "+str(nfB)+", "+str(nmA)+", "+str(nmB)+"\n"
for t in range(1,czas+1):
n=0
pr=1/(1+exp(-(ar*N+br)))
ps=1/(1+exp(-(ass*N+bs)))
for i in range(N):
osob=populacja[i]
if osob.PLEC=='F' and random()<pr :
potom=stanosobnika()
potom.NR=ostnr+1
ostnr=ostnr+1
if random()<0.5:
potom.PLEC='F'
else:
potom.PLEC='M'
if random()<0.5:
potom.GEN1=osob.GEN1
else:
potom.GEN1=osob.GEN2
ojciec=populacja[randrange(N)]
if random()<0.5:
potom.GEN2=ojciec.GEN1
else:
potom.GEN2=ojciec.GEN2
populacja1.append(potom)
n=n+1
if random()<ps:
pass
else:
populacja1.append(osob)
n=n+1
N=n
if N>100000:
break
Nf=0
Nm=0
NfAA=0
NfAB=0
NfBB=0
NmAA=0
NmAB=0
NmBB=0
nfA=0
nfB=0
nmA=0
nmB=0
for i in range(N):
osob=populacja1[i]
if (osob.PLEC=='F' and osob.GEN1=='A' and osob.GEN2=='A'):
NfAA=NfAA+1
nfA=nfA+2
if (osob.PLEC=='F' and osob.GEN1=='A' and osob.GEN2=='B'):
NfAB=NfAB+1
nfA=nfA+1
nfB=nfB+1
if (osob.PLEC=='F' and osob.GEN1=='B' and osob.GEN2=='A'):
NfAB=NfAB+1
nfA=nfA+1
nfB=nfB+1
if (osob.PLEC=='F' and osob.GEN1=='B' and osob.GEN2=='B'):
NfBB=NfBB+1
nfB=nfB+2
if (osob.PLEC=='M' and osob.GEN1=='A' and osob.GEN2=='A'):
NmAA=NmAA+1
nmA=nmA+2
if (osob.PLEC=='M' and osob.GEN1=='A' and osob.GEN2=='B'):
NmAB=NmAB+1
nmA=nmA+1
nmB=nmB+1
if (osob.PLEC=='M' and osob.GEN1=='B' and osob.GEN2=='A'):
NmAB=NmAB+1
nmA=nmA+1
nmB=nmB+1
if (osob.PLEC=='M' and osob.GEN1=='B' and osob.GEN2=='B'):
NmBB=NmBB+1
nmB=nmB+2
Nf = NfAA + NfAB + NfBB
Nm = NmAA + NmAB + NmBB
populacja=populacja1
populacja1=[]
tekst=tekst+str(pow)+", "+str(t)+", "+str(N)+", "+str(Nf)+", "+str(Nm)+", "+str(NfAA)+", "+str(NfAB)
tekst=tekst+", "+str(NfBB)+", "+str(NmAA)+", "+str(NmAB)+", "+str(NmBB)+", "+str(nfA)
tekst=tekst+", "+str(nfB)+", "+str(nmA)+", "+str(nmB) +"\n"
populacja=[]
open('wyniki.txt', 'a').write(tekst)
wyniki.close()
|
Specyficzne strategie rozrodcze realizowane są w populacjach osobników tri- i tetraplodalnych. Jest to jednak strategia dość rzadka, a jednocześnie obejmująca potencjalnie kilka rodzajów dziedziczenia genów. Zostawiam budowę takich symulatorów specjalistom od tego typu roślin i innych tego typu organizmów. Widać jednak, że jest to możliwe i nietrudne.
Analiza wyników symulacji
Wyliczenie w każdym kroku czasowym liczby osobników o różnych genotypach AA, AB i BB pozwala na określenie struktury genotypowej populacji i jej zmiany w czasie. Można ją narysować dla każdego powtórzenia tworząc szereg wykresów słupkowych pokazujących na dole udział procentowy osobników o genotypie AA, w środku udział procentowy osobników o genotypie AB a na górze – udział procentowy osobników o genotypie BB. Natomiast gdy tak samo pokaże się zmiany w czasie udziału procentowego genów o allelu A w stosunku do udziału procentowego allelu B to Wykres taki obrazuje zmiany w czasie struktury genetycznej. Dla populacji klonalnej (haploidalnej, bo taka zaprogramowano) struktura genotypowa i genetyczna są takie same.
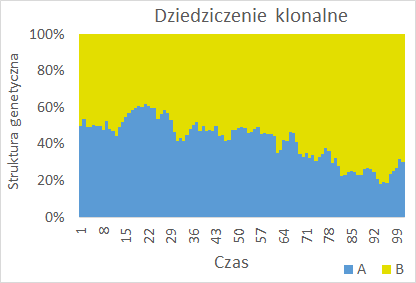
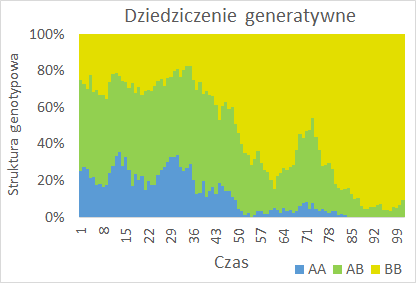
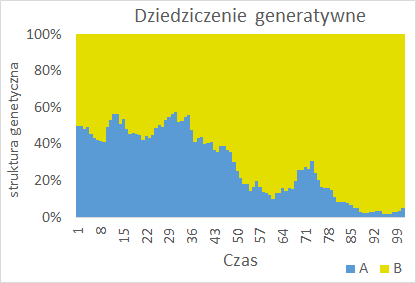
Jest to obraz uzyskiwany dla badań pojedynczych populacji. Widać na nim dryf genetyczny – losowe zmiany struktury genetycznej, które zazwyczaj dość szybko powodują, populacje stają się monomorficzne. Wykonany model populacji jest zgodny z modelem dryfu genetycznego wprowadzonym do nauki przez Sewalla Wrighta w 1921 roku.
Wyciąganie wniosków dotyczących kierunku i tempa dryfu genetycznego na podstawie wykresów zrobionych dla jednej populacji nie jest możliwe. Należy zatem zobrazować zjawisko dla wielu powtórzeń symulacji. Gdy porównujemy ze sobą wiele populacji najlepiej jest operować frakcją jednego z alleli, czyli liczbą:
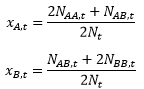
gdzie N jest liczebnością populacji w danym kroku czasowym, NAA – liczbą homozygot AA, NAB – liczbą heterozygot AB, NBB – liczbą homozygot BB, NA liczbą genów o allelu A w puli genowej i NB liczba genów o allelu B w puli genowej populacji. Można wtedy na jednym wykresie pokazać uzyskane w wyniku symulacji zmiany w czasie frakcji allelu A dla wielu powtórzeń symulacji. Ponieważ w przypadku dwóch alleli zawsze XB=1-XA wystarczy obrazować w czasie zmiany frakcji jednego z alleli, np. allelu A.
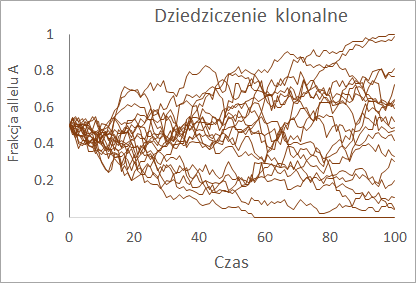
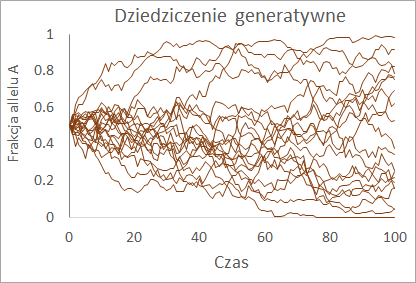
Ostatnie wykresy są typowe dla procesów niestabilnych. Aby jednak ocenić stabilność struktury genetycznej populacji należy wyliczyć wartości przewidywane dla zmian w czasie struktury genetycznej i ocenić jej stabilność.
Przewidywana struktura genetyczna populacji klonalnej
Przy porównaniu dwóch kolejnych pokoleń populacji rozmnażającej się klonalnie jak i populacji o rozrodzie generatywnym można wykonać rozważania matematyczne. W populacji klonalnej jeżeli w chwili t żyje NA,t osobników o genie o allelu A i NB,t osobników o genie o allelu B to prawdopodobieństwa, że rozrodzi się RA osobników spośród NA,t oraz RB osobników spośród NB,t są równe:
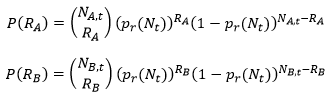
gdzie N=NA+NB. Podobnie prawdopodobieństwa, że Umrze się SA osobników spośród NA oraz SB osobników spośród NB są równe:
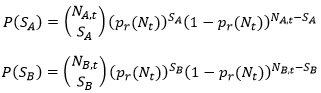
Wychodząc z tych wzorów można utworzyć macierz stochastyczną dla tego procesu, wyliczyć warunkowe wartości oczekiwane i ostatecznie wyprowadzić wzory na przewidywane zmiany frakcji alleli z pokolenia na pokolenie. Jednak wzory te wyprowadza się prościej, bez potrzeby definiowania prawdopodobieństwa w macierzy stochastycznej. Wartości przewidywane liczby osobników rozradzających się z genem o danym allelu i wartości oczekiwanej liczby osobników zmarłych z genem o danym allelu dane są wzorami:
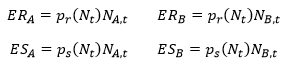
Wynika stąd, że:
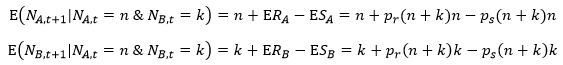
Są wzory na warunkowe wartości oczekiwane liczby osobników z genem o danym allelu. Wynikają z nich wzory na wartości przewidywane tych wielkości:
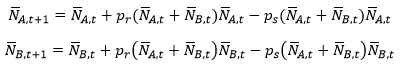
Z tego wzoru wynika, że przewidywana frakcja allelu A tworzy ciągi rekurencyjny:
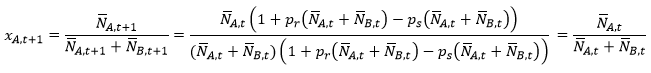
Ostatecznie dla każdego t: 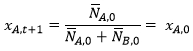
Czyli frakcja allelu A nie zmienia się w czasie i jest równa frakcji tego allelu w populacji początkowej. Jest stanem równowagi, ale żadne inna krzywa przewidywana wyprowadzona z innej frakcji początkowej allelu A nie zmierza do niego. To z biologicznego punktu widzenia oznacza niestabilność przewidywanych frakcji allelu A. Oczywiście dotyczy to także frakcji allelu B.
Przewidywana struktura genetyczna populacji nierozdzielnopłciowej z rozrodem generatywnym
Wyprowadzimy teraz wzory na przewidywane dynamiki liczebności osobników o danym genotypie w populacji o rozrodzie generatywnym. Na początku należy zauważyć, że losowanie z populacji osobnika, a następnie losowanie jednego z jego dwóch genów jest równoważne losowaniu jednego genu z puli genowej populacji. Równoważność polega na tym, że przy rozważaniu podwójnego losowania (osobnika i jego genu) i przy losowaniu genu z puli genowej powstają dokładnie te same przestrzenie probabilistyczne.
Jeżeli w chwili t w populacji jest NAA,t osobników o genotypie AA, NAB,t osobników o genotypie AB i NBB,t osobników o genotypie BB to w puli genowej populacji jest 2NAA,t+NAB,t genów o allelu A i NAB,t+2NBB,t genów o allelu B. Prawdopodobieństwa wylosowania genu o określonym alleu są równe:
![]()
Prawdopodobieństwo, że potomek będzie miał określony genotyp jest równe prawdopodobieństwu dwukrotnego losowania jakiegoś genu (ze zwracaniem – geny po wylosowaniu nie przechodzą do potomka tylko się kopiują). Prawdopodobieństwa, że potomek będzie miał genotyp AA jest zatem równe (xA)2, że będzie miał genotyp AB jest równe: 2(xA)(xA) (jako że rozważamy losowanie pierwszego genu A i drugiego B plus losowanie pierwszego genu B i drugiego A) oraz, że będzie miał genotyp BB będzie równe (xBB)2.
Przy założeniu że ogólna liczba potomków wynosi R możemy wyliczać prawdopodobieństwa powstania określonych genotypów wśród potomków za pomocą wzorów dwumianowych:
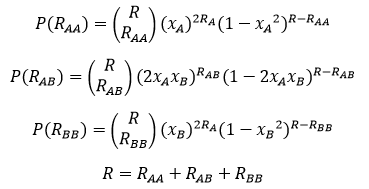 Dla każdej możliwej liczby R rozradzających się osobników (równej liczbie potomków) wartości oczekiwane liczb potomków o danym genotypie są równe (xA)2R, 2(xA)(xA)R, (xBB)2R, a wartość oczekiwana wszystkich potomków wynikająca z mechanizmu regulacyjnego wynosi ER=pr(Nt)Nt. Wynikają stąd następujące wzory na warunkowe wartości oczekiwane liczby osobników o danych genotypach w populacji:
Dla każdej możliwej liczby R rozradzających się osobników (równej liczbie potomków) wartości oczekiwane liczb potomków o danym genotypie są równe (xA)2R, 2(xA)(xA)R, (xBB)2R, a wartość oczekiwana wszystkich potomków wynikająca z mechanizmu regulacyjnego wynosi ER=pr(Nt)Nt. Wynikają stąd następujące wzory na warunkowe wartości oczekiwane liczby osobników o danych genotypach w populacji:
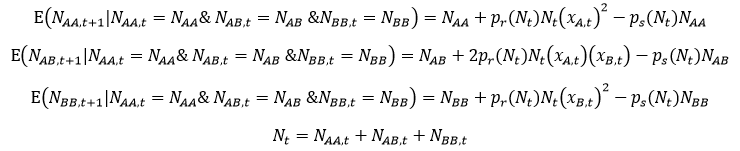
Stąd możemy wyprowadzić wzory na przewidywane liczby osobników o danych genotypach w kolejnych krokach czasowych:
Sumując stronami trzy pierwsze równania otrzymujemy:
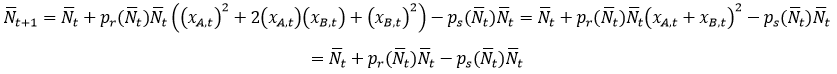
Wyprowadzimy teraz wzór na przewidywane frakcje allelu A w kolejnych krokach czasowych:
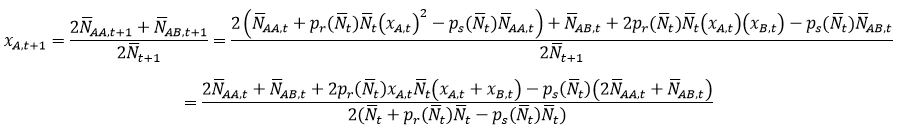
Ponieważ xA,t+xB,t=1 oraz 2NAA,t+NAN,t=2xA,tNt to:
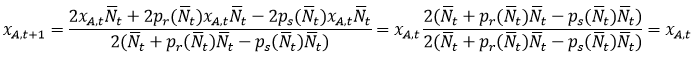
Co oznacza , że nie przewidujemy zmian w czasie frakcji allelu A. Ostatecznie xA,t=xA,0 dla dowolnego czasu t. Wskazuje to na niestabilność struktury genetycznej populacji z dziedziczeniem Mendlowskim, co uwidacznia się na wykresie.
Przewidywana struktura genetyczna populacji rozdzielnopłciowej o rozrodzie generatywnym
Dla populacji rozdzielnopłciowej należy rozpatrywać osobno frakcje genów A i B u samic (z indeksem f) i u samców (z indeksem m). Wyliczyć je można ze wzorów:
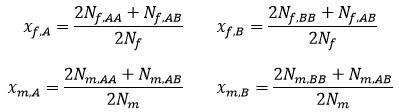
Frakcje te są równe prawdopodobieństwom wylosowanie allelu A albo B z puli genowej samic oraz prawdopodobieństwu wylosowania alleli A albo B z puli genowej samców. Losowanie określonych genów dla potomka od samicy i od samca są zdarzeniami niezależnymi. Zatem:
- Prawdopodobieństwo pojawienia się samicy o genotypie AA wśród nowonarodzonych samic jest równe:
xf,A·xm,A - Prawdopodobieństwo pojawienia się samicy o genotypie AB wśród nowonarodzonych samic jest równe:
xf,A·xm,B + xf,B·xm,A - Prawdopodobieństwo pojawienia się samicy o genotypie BB wśród nowonarodzonych samic jest równe:
xf,B·xm,B - Prawdopodobieństwo pojawienia się samca o genotypie AA wśród nowonarodzonych samców jest równe:
xf,A·xm,A - Prawdopodobieństwo pojawienia się samca o genotypie AB wśród nowonarodzonych samców jest równe:
xf,A·xm,B + xf,B·xm,A - Prawdopodobieństwo pojawienia się samca o genotypie BB wśród nowonarodzonych samców jest równe:
xf,B·xm,B
Po rozważaniach probabilistycznych (prostych, ale ze względu na ich 6-krotne powtarzanie po prostu nudnych) uzyskamy następujące wzory na przewidywane liczebności samic i samców o określonych genotypach:

Tylko 6 pierwszych równań tworzy 6-wymiarowy ciąg rekurencyjny będący przewidywaną liczebności samców i samic o określonych genotypach. Zaznaczone na niebiesko równania mają charakter nie tylko podsumowujący uzyskane wielkości, ale możliwość generowania kolejnych wartości tego wektora. Występują one bowiem we wzorach generujących 6 pierwszych wielkości. Gdyby je wszystkie wstawić w odpowiednie miejsca powstałyby bardzo długie formuły i trudno byłoby zapanować na ewentualnymi błędami.
Jeżeli chodzi o zmiany frakcji allelu A z pokolenia na pokolenie, to można dowieść tylko, że:
![]()
Jednocześnie frakcje dla samic i samców będą zmierzały w czasie do zrównania się. Jeżeli w puli genowej populacji początkowej frakcja alleli A u samic była większa niż u samców, to zaobserwujemy w czasie jej spadek u samic i wzrost u samców. Nadal jednak jest to układ niestabilny bo każda zmiana frakcji tego allelu w populacji początkowej spowoduje przesunięcie się obu ciągu opisującego frakcje alleli samic oraz samców. Nie będzie wśród nich atraktorów.
Prawo Hardy’ego Weinberga
Przy dziedziczeniu Mendlowskim wzory na przewidywane liczby osobników o określonych genotypach AA, AB, i BB w zależności od frakcji alleli A i B w puli genowe w poprzednim pokoleniu wyglądają prawie dokładnie tak samo jak prawo Hardy’ego-Weinberga, podstawowe prawo genetyki populacyjnej, z którym studenci ekologii spotkają się wielokrotnie.
| PRAWO HARDY’EGO-WEINBERGA
Zakładamy, że w populacji występują takie czynniki jak:
W takiej populacji, jeżeli w puli genowej rodziców frakcje genów o allelach A i B wynoszą xA i xB odpowiednio, to frakcje potomków o genotypach:
|
Wszystko co należy zrobić, to utożsamić prawdopodobieństwo z frakcją. Dla wielu osób jest to tak oczywiste, że nie widzą potrzeby rozważań probabilistycznych. Jednak utożsamienie prawdopodobieństwa z frakcją jest możliwe tylko, gdy liczebność populacji jest nieskończona.
Liczby osobników rozradzających się i zmarłych losowane są w tych programach z rozkłady Bernoulliego. Z im większej populacji są one losowane tym wydają się być bliższe wartości całego rozkładu równej pN – co widać na wykresach rozkładów dwumianowych dla p=0.4 i N=10, 100, 1000 i 10000:
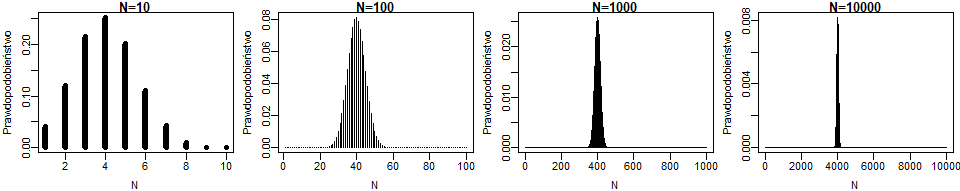
Przy wzroście liczby potomków R prawdopodobieństwa w rozkładzie dwumianowym coraz bardziej koncentrują się na wartościach oczekiwanych tego rozkładu. Nazywamy to zjawisko prawem wielkich liczb Bernoulliego. Można więc oczekiwać, że przy dużych liczbach potomków zmiany struktury genetycznej i frakcji alleli będą coraz wolniejsze. Wielokrotnie potwierdzono to dokumentalnie zarówno w modelach jak i dla prawdziwych populacji.
Dla populacji rozdzielnopłciowej odpowiednikiem prawa Hardy’ego-Weinberga jest następujące twierdzenie:
| PRAWO HARDY’EGO-WEINBERGA W POPULACJI ROZDZIELNOPŁCIOWEJ DLA GENÓW NIE ZWIĄZANYCH Z DETERMINACJĄ PŁCIZakładamy, że w populacji występują takie czynniki jak:
W takiej populacji, jeżeli w puli genowej rodziców frakcje genów samic o allelach A i B wynoszą xf,A i xf,B odpowiednio oraz frakcje genów samców o allelach A i B wynoszą xm,A i xm,B odpowiednio, to frakcje potomków o genotypach:
Wśród tych potomków w każdej grupie jest taka sama proporcja samic/samców, jaka jest założona lub obserwowana jako częstość narodzin samicy/samca w całej populacji. |
Zadania dla studentów
Dryf generyczny zobaczymy uruchamiajac programy w Pythonie, których kody podane zostały w tym rozdziale. Dla tych samych parametrów wyliczymy ciagi wartości przewidywanych, co najłatwiej jest wykonać w arkuszu kalkulacyjnym Excel lub Calc. Poniezej podano przepisy ta takie wyliczenia z mozliwoscią szybkiej zamiany parametrów modelu.
Model populacji rozmnażającej się klonalnie
Otworzyć pusty plik arkusza kalkulacyjnego. W polach A1, A2, A3, A4 wpisać nazwy parametrów modelu (ar, br, as, bs), a w polach B1, B2, B3, B4 wymyślone przez siebie wartości tych parametrów. Mają być takie, aby populacja była stabilna i cechowała się stanem równowagi równym 100 (mogą być takie jak w podanym programie).
W polu C1 wpisać “czas” a w polach C2:C502 liczby od 0 do 500.
W polu D1 wpisać “pr”. W polu E1 wpisać “ps”. W polach F1 i G1 wpisać “NA” i “NB” odpowiednio. Będą w nich wyliczane liczby osobników z genem o danym allelu.
W polach F2 i G2 wpisać liczebności początkowe osobników o danym genotypie.
W polu D3 należy wpisać formułę =1/(1+exp(-($B$1*(F2+G2)+$B$2)))
W polu E3 należy wpisać formułę =1/(1+exp(-($B$3*(F2+G2)+$B$4)))
W polu F3 należy wpisać formułę =F2+D3*F2-E3*F2
W polu G3 należy wpisać formułę =G2+D3*G2-E3*G2
Zaznaczyć formuły D3:G3 i przeciągnąć w dół do wiersza 502.
W polu H1 wpisać frakcja A. W polu H2 wpisać formułę =F2/(F2+G2). Zaznaczyć pole i przeciągnąć w dół do wiersza 502.
Zobrazować przewidywaną strukturę genetyczną=genotypową populacji stosując wykres słupkowy z pionowym rozkładem procentowym. Podpisać osie. Co się dzieje jak zmieniamy liczebności początkowe? W oparciu o poprzedni referat powiedzieć jaki typ stabilności pokazuje taki układ (wszystkie krzywe są stanami równowagi, ale nie ma powrotów do stanu równowagi).
Zobrazować na jednym wykresie zmiany w czasie frakcji allelu A dla różnych liczebności początkowych NA i NB. Jaki typ stabilności cechuję tę zmienną?
Uruchomić pierwszy z podanych programów zmieniając mu wartości ar, br, ass i bs na takie same, jakie zastosowano w arkuszu kalkulacyjnym i wstawiając za czas liczbę 500.
wyniki symulacji przenieść do arkusza kalkulacyjnego, tak by znalazły się one w 5 kolumnach. Skopiować je do wcześniej wykonanego pliku.
Zobrazować dynamiki liczebności populacji.
Do wykresu zmian w czasie proporcji osobników z genami o różnych allelach dodać zmiany w czasie proporcji osobników NA i NB (w formie nie słupkowej a liniowej) wyliczone dla pierwszego powtórzenia symulacji.
Wyliczyć zmiany frakcji allelu A we wszystkich 20 powtórzeniach.
Dodać wykres zależności frakcji allelu A od czasu do drugiego wcześniej wykonanego wykresu.
Wykresy zapamiętać.
Sprawdzić czy zauważone zależności będą takie same, gdy liczebność populacji zostanie wielokrotnie zwiększona. W tym celu można skopiować cały plik Excela/Calca by oszczędzić sobie pracy.
Zmienić ar i as dzieląc je przez 10 (czyli gdy było ar=-0.005 to teraz będzie ar=-0.0005; podobnie as=0.005 zmieni się na as=0.0005).
Wykonać symulację z nowymi parametrami i N0=1000. Wyniki otworzyć w Excelu i podmienić nimi stare wyniki metodą kopiuj-wklej. Powinno to natychmiast zaktualizować wykresy. Zapamiętać.
Model populacji nierozdzielnopłciowej rozmnażającej się generatywnie
Otworzyć pusty plik Excela lub Calca. W polach A1, A2, A3, A4 wpisać nazwy parametrów modelu (as, bs, ar, br), a w polach B1, B2, B3, B4 wymyślone przez siebie wartości tych parametrów. Mają być takie, aby populacja była stabilna i cechowała się stanem równowagi bliskim liczbie 100 (mogą być takie jak w podanym programie).
W polu C1 wpisać “czas” a w polach C2:C502 liczby od 0 do 500.
W polu D1 wpisać “ps”. W polu E1 wpisać “pr”. W polach F1, G1 i H1 wpisać “NAA”, “NAB” i “NBB” odpowiednio. Będą w nich wyliczane liczby osobników o danych genotypach. W pola I1 i J1 wpisać “xA” i “xB”. Tu będą wyliczane frakcje genów A i genów B w puli genowej populacji.
W polach F2, G2 i H2 wpisać liczebności początkowe osobników o danym genotypie.
W polu I2 wyliczyć frakcję genów A w puli genowej. W polu J2 wyliczyć frakcje genów B w puli genowej. Przypominam, że frakcje te wyliczane są wg. wzoru:
![]()
Należy samemu napisać odpowiednią formułę po znaku = odwołując się do komórek F2, G2 i H2.
W polu D3 należy wpisać formułę =1/(1+exp(-($B$1*(F2+G2+H2)+$B$2)))
W polu E3 należy wpisać formułę =1/(1+exp(-($B$3*(F2+G2+H2)+$B$4)))
W polu F3 należy wpisać formułę =F2+E3*(F2+G2+H2)*I2^2-D3*F2
W polu G3 należy wpisać formułę =G2+E3*(F2+G2+H2)*2*I2*J2-D3*G2
W polu H3 należy wpisać formułę =H2+E3*(F2+G2+H2)*J2^2-D3*H2
Formuły z pół I2 i J2 przeciągnąć do I3 i J3. Zaznaczyć pola od D3 do J3 i przeciągnąć wszystkie formuły do wiersza 502.
Wykonać wykres przewidywanej struktury genotypowej populacji. Zatytułować podpisać osie, wprowadzić legendę.
Wykonać wykres frakcji allelu A (kolumny I). Frakcja allelu B to 1-frakcja allelu A i nie trzeba jej rysować. Zatytułować, podpisać osie, legendę.
Zapamiętać.
Uruchomić program symulacyjny, którego kod został wcześniej podany i wprowadzić takie same parametry jakie ustalono w pliku excelowym, 500 kroków czasowych. Plik wynikowy otworzyć w arkuszu kalkulacyjnym, tak by wyniki znalazły się 8 kolumnach.
Skopiować te dane do wcześniej wykonanego pliku Excelowego/Calcowego.
Wykonać wykres dynamik liczebności symulowanych populacji. Zatytułować, podpisać osie.
Wykonać wykres liniowy zmian w czasie proporcji osobników NAA, NAB i NBB dla pierwszego powtórzenia symulacji. Wybrać opcję wykres warstwowy. Wykres taki nałożyć na wykonany wcześniej wykres zmian w czasie struktury genotypowej populacji.
Wykonać jeden wykres zależności frakcji allelu A od czasu dla wszystkich 20 powtórzeń i dołożyć do drugiego wykresu wykonanego wcześniej.
Sprawdzić czy zauważone zależności będą takie same, gdy liczebność populacji zostanie wielokrotnie zwiększona. W tym celu można skopiować plik Excela/Calca by oszczędzić sobie pracy.
Zmienić ar i as dzieląc je przez 10 (czyli gdy było ar=-0.005 to teraz będzie ar=-0.0005; podobnie as=0.005 zmieni się na as=0.0005).
Wykonać symulację z nowymi parametrami i N0=1000. Wyniki otworzyć w Excelu i podmienić nimi stare wyniki metodą kopiuj-wklej. Powinno to natychmiast zaktualizować wykresy. Zapamiętać.
- Czy przewidywane zmiany liczb osobników o określonych genotypach pozwalają na przewidywanie wyników symulacji?
- Czy brak zmian dla przewidywanych frakcji allelu A w czasie można skomentować, że równie często dryf genetyczny idzie w górę jak i w dół i średnio wychodzi na brak zmian?
- Czy wzrost liczebności populacji powoduje zwolnienie dryfu genetycznego jak to założono w teorii?
- Czy gdy liczebność populacji wzrośnie do nieskończoności do dryf zniknie?
- Z prawa Hardy’ego Weinberga wynika, że frakcje alleli nie zmieniają się w czasie. Dla kreacjonistów do dowód na brak ewolucji. Ustosunkuj się do tego rozumowania.
- Czy prawo Hardy’ego-Weinberga jest zaprzeczeniem dryfu genetycznego czy jego uzupełnieniem?
Model populacji rozdzielnopłciowej rozmnażającej się generatywnie
Otworzyć pusty plik Excela lub Calca. W polach A1, A2, A3, A4 wpisać nazwy parametrów modelu (as, bs, ar, br), a w polach B1, B2, B3, B4 odpowiednie wymyślone wartości tych parametrów (ale takie, aby populacja była stabilna i NE=100).
W polu C1 wpisać “czas” a w polach C2:C502 liczby od 0 do 500.
W polu D1 wpisać “ps”. W polu E1 wpisać “pr”. W polach F1, G1 i H1 wpisać “NfAA”, “NfAB” i “NfBB” odpowiednio. W polu E1 wpisać “pr”. W polach I1, J1 i K1 wpisać “NmAA”, “NmAB” i “NmBB” odpowiednio. Będą w nich wyliczane liczby osobników o danej płci i danych genotypach. W pola L1 i M1 wpisać “xfA” i “xfB”. W pola N1 i O1 wpisać “xmA” i “xmB”. Tu będą wyliczane frakcje genów A i genów B w puli genowej populacji.
W polach F2, G2, H2, I2, J2 i K2 wpisać liczebności początkowe osobników o danej płci i danym genotypie (20, 20, 10, 10, 20, 20).
W polach L2, M2, N2 i O2 wyliczyć frakcję genów samic i samców w puli genowej. Wzory na te frakcje podano na początku ostatniego podrozdziału. Należy samemu napisać odpowiednią formułę po znaku = odwołując się do komórek F2, G2, H2, I2, J2 i K2.
W polu D3 należy wpisać formułę =1/(1+exp(-($B$1*(F2+G2+H2+I2+J2+K2)+$B$2))).
W polu E3 należy wpisać formułę =1/(1+exp(-($B$3*(F2+G2+H2+I2+J2+K2)+$B$4))).
W polu F3 należy wpisać formułę =F2+0.5*E3*(F2+G2+H2)*N2*L2-D3*F2
W polu G3 należy wpisać formułę =G2+0.5*E3*(F2+G2+H2)*(N2*M2+O2*L2)-D3*G2
W polu H3 należy wpisać formułę =H2+0.5*E3*(F2+G2+H2)*O2*M2-D3*H2
W polu I3 należy wpisać formułę =I2+0.5*E3*(F2+G2+H2)*N2*L2-D3*I2
W polu J3 należy wpisać formułę =J2+0.5*E3*(F2+G2+H2)*(N2*M2+O2*L2)-D3*J2
W polu K3 należy wpisać formułę =K2+0.5*E3*(F2+G2+H2)*O2*M2-D3*K2
Formuły z pół L2, M2, N2 i O2 przeciągnąć do L3:O3. Zaznaczyć pola od D3 do O3 i przeciągnąć wszystkie formuły do wiersza 502.
Wykonać wykres przewidywanych liczebności samic i samców o genotypach AA, AB i BB (czas i kolumny F, G, H, I, J, K; wykres punktowy, połączony liniami. Zatytułować, opisać osie. Wprowadzić legendę z oznaczeniem nazw płci i genotypów. Nazwać plik zadanie21.xls i zapamiętać.
Wykonać wykres przewidywanych frakcji allelu A samic i samców. Podpisać osie, zatytułować.
Uruchomić program symulacyjny, którego kod został wcześniej podany i wprowadzić takie same parametry jakie ustalone zostały w Excelu, 500 kroków czasowych, jedno powtórzenie.
Powstały plik INFODYN.TXT otworzyć w Excelu i skopiować do zadanie21.xls.
Dodać liczebności samic i samców danych genotypów do wykonanego wcześniej wykresu dynamik. Nadać im ten sam kolor, co odpowiednie przewidywane liczebności. Zapamiętać.
Dodać frakcje alleli A samic i samców do drugiego wykresu. Nadać im te kolory, które mają odpowiednie przewidywane frakcje. Poprawić tytuł i legendę.
Trzeci i czwarty wykres należy wykonać tylko dla symulowanych danych. Frakcje danego allelu w puli genowej pokazuje się na ogół w postaci stojących na sobie słupków o dwóch kolorach. Dryf genetyczny obrazuje się zatem jako stojące obok siebie takie słupki.
Należy zaznaczyć kolumny z frakcją allelu A i frakcją allelu B samic, kliknąć na ikonkę wykresy, wybrać kolumnowy (słupki pionowe) i drugi lub trzeci podtyp (stojące na sobie słupki).
Na zapewne całkiem czarnym wykresie trzeba zmienić sposób rysowania słupka. Kliknąć dwukrotnie na dolną część słupków, tak aby pojawiło się okienko ich formatowania i w części “obramowanie” wybrać brak. Analogicznie postąpić z górną częścią słupków.
Podpisać osie, zatytułować poprawić legendę.
Analogiczny wykres zrobić dla samców. Czy wykresy te są podobne?
W populacji rozdzielnopłciowej stabilna jest liczebność i struktura płciowa, a struktura genetyczna jest niestabilna. Stąd obserwowany jest dryf genetyczny. Wykonany model tłumaczy szybkość zmian struktury genetycznej rzeczywistych populacji. Dryf genetyczny, jak się wydaje, jest motorem nieselektywnej ewolucji (zanikanie różnorodności genetycznej w niedużych izolowanych populacjach, pozostawanie u wszystkich osobników jednej przypadkowej cechy charakteryzującej któregoś z przodków).